|
|

|

|
| e-Pub |
Section: New Results
Meristem functioning and development
In axis 2 work focuses on the creation of a virtual meristem, at cell resolution, able to integrate the recent results in developmental biology and to simulate the feedback loops between physiology and growth. The approach is subdivided into several sub-areas of research.
Data acquisition and design of meristem models
-
Improvement of the MARS-ALT pipeline robustness Meristem, laser microscopy, image reconstruction, cell segmentation, automatic lineaging
Participants : Léo Guignard, Christophe Godin, Christophe Pradal, Grégoire Malandain [Morpheme, Inria] , Gaël Michelin [Morpheme, IPL Morphogenetics, Inria] , Guillaume Baty, Sophie Ribes [IBC, UM] , Jan Traas [RDP, ENS] , Patrick Lemaire [CRBM, CNRS] , Yassin Refahi [RDP, ENS-Lyon / Sainsbury Lab, Cambridge, UK] .
This research theme is supported by a PhD FRM grant, Jan Traas's ERC, Inria ADT programme and the Morphogenetics Inria Project Lab.
The MARS-ALT (Multi-Angles Registration and Segmentation - Automatic Lineage Tracking) software pipeline [6] automatically performs a segmentation at cell resolution from 3D or 2D voxel images where the membranes/walls are marked (by a die for example) and makes it possible to follow the lineage of these cells through time.
This year, the ALT tracking pipeline has been reformulated by using a generic cell modeling approach (enabling for example more than one cell division), and both stability and robustness were improved. The modeling approach is generic and can be used on other kind of data (nuclei, human cells, ...). These trials will be conducted during the year. Moreover, the architecture of the image processing components has been modified (plugin approach) and integrated with the TissueLab platform. Some vizualisation tools have been improved, and the platform includes a module allowing an interaction with data (Alizon Konig, master internship). This point enables an efficient creation of gold standard to validate segmentation results.
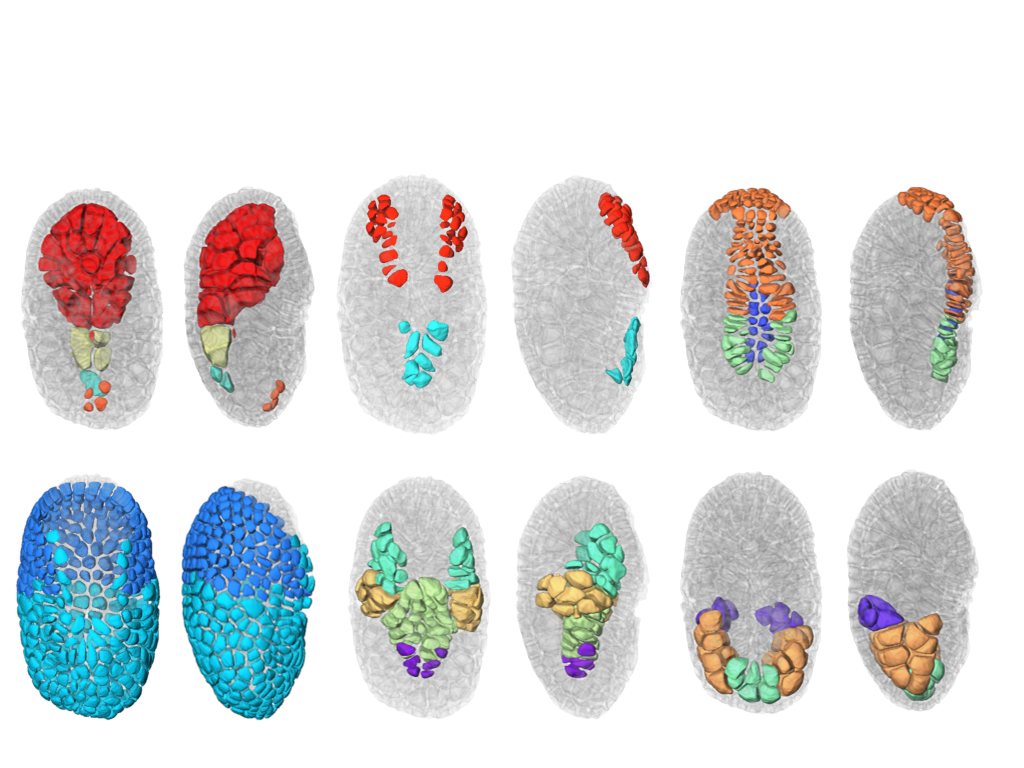
This year, we also finalize the development of a a new segmentation and tracking pipeline, ASTEC (Adaptive Segmentation and Tracking of Embryonic Cells). ASTEC is a one-pass algorithm (in contrast to MARS-ALT, that perform first the segmentation and then the tracking in two-passes) that is best suited for movies with numerous close time-points acquired at high spatio-temporal resolution. This pipeline takes advantage of information redundancy across the movies and biological knowledge on the segmented organism to constrain and improve the segmentation and the tracking. We used this one-pass algorithm to segment and track all cell shapes of a developing embryo of the marine invertebrate Phallusia mammillata. As a result we obtained the full track of the shapes of all the cells from the 64 cell stage up to the early tailbud stage (1030 cells undergoing 640 division events followed across 180 time-points through 6 hours of development imaged every 2 minutes, Figure 2 ).
Based on this quantitative digital representation, we systematically identified cell fate specification events up to the late gastrula stage. Computational simulations revealed that remarkably simple rules integrating measured cell-cell contact areas with spatio-temporal expression data for extracellular signalling molecules are sufficient to explain most early cell inductions. This work suggests that in embryos developing with stereotyped cell shapes and positions (like Phallusia mammillata embryos), the genomic constraints for precise gene expression levels are relaxed, thereby allowing rapid genome evolution.
-
Creating mesh representation of cellular structures
Participants : Guillaume Cerutti, Sophie Ribes, Christophe Godin, Géraldine Brunoud [RDP, ENS] , Carlos Galvan-Ampudia [RDP, ENS] , Teva Vernoux [RDP, ENS] , Yassin Refahi [RDP, ENS, Sainsbury Lab] .
This research theme is supported the HFSP project Biosensors.
To produce a more efficient data structure accounting for the geometry of cellular tissues, we studied the problem of reconstructing a mesh representation of cells in a complex, multi-layered tissue structure, based either on membrane/wall images segmented using MARS or on nuclei images of shoot apical meristems. The construction of such mesh structures for plant tissues is currently a missing step in the existing image analysis pipelines.
We developed tools to reconstruct a 3D cell complex representing the tissue, based on the dual simplicial complex of cell adjacencies. This set of tetrahedra is optimized from a reasonable initial guess to match the adjacencies in the tissue, which proved to produce a very faithful reconstruction[39] . We also developed a set of methods to triangulate such reconstructions, and enhance the quality of triangular mesh representations of plant tissue, simultaneously along several criteria [28] .
These tools can produce light discrete representations of the cell interfaces that enables fast visualization, information projection, and quantitative analysis of the tissue, and have given way to to some of the first biomechanical simulations on real-world data.
-
Design of 3D digital atlases of tissue development
Participants : Sophie Ribes, Yassin Refahi [RDP, ENS, Sainsbury Lab] , Guillaume Cerutti, Christophe Godin, Christophe Pradal, Christophe Pradal, Frédéric Boudon, Gregoire Malandain [RDP, ENS] , Gaël Michelin [RDP, ENS] , Guillaume Baty, Jan Traas [RDP, ENS] , Teva Vernoux [RDP, ENS] , Patrick Lemaire [CRBM, CNRS] , Françoise Monéger [RDP, ENS] .
This research theme is supported the Inria Project Lab Morphogenetics, the ADT Mars-Alt and the HFSP project Biosensors.
To organize the various genetic, physiological, physical, temporal and positional informations, we build a spatialized and dynamic database [67] . This database makes it possible to store all the collected information on a virtual 3D structure representing a typical organ. Each piece of information has to be located spatially and temporally in the database. Tools to visually retrieve and manipulate the information, quantitatively through space and time are being developed. For this, the 3D structure of a typical organ has been created at the different stages of development of the flower bud. This virtual structure contains spatial and temporal information on mean cell numbers, cell size, cell lineages, possible cell polarization (transporters, microtubules), and gene expression patterns. Such 3D digital atlas is mainly descriptive. However, like for classical databases, specific tools make it possible to explore the digital atlas according to main index keys, in particular spatial and temporal keys. Both a dedicated language and a 3D user interface are being designed to investigate and query the 3D virtual atlas. Current developments of this tool consist in using directly the segmented images produced from laser microscopy to build the atlas. To better represent the development of a biological population, a method to compute an "average" structure is investigated.

Shape analysis of meristems
Participants : Jonathan Legrand, Pierre Fernique, Frédéric Boudon, Yann Guédon, Christophe Godin, Pradeep Das [RDP, ENS] , Arezki Boudaoud [RDP, ENS] .
At cellular resolution, we studied the organization of cells in the meristems. The MARS-ALT pipeline provides rich spatio-temporal data sets for analyzing the development of meristems. A first step consisted of designing a dedicated graph structure for efficiently representing the spatial (adjacency between cells) and temporal (cell division) relationships between cells. Various variables can be attached either to the vertices (e.g. cell volume, inertia axes) or the edges (e.g. wall surface, distance between cell centroids). This graph may be augmented by new variables resulting from various spatial or temporal filtering (e.g. cell volumetric growth). Looking at homogeneous regions in the variable value space, cellular patterns can be identified.
Considering the highly-structured nature of our data (time and space structuring) and the potential diversity and heterogeneity of possible cell descriptors, we developed two complementary approaches:
-
A first one that favours the spatial structuring: In this approach, the cell neighbourhood and the cell descriptors are jointly taken into account in a clustering approach whose objective is to identify a small number of clusters corresponding to well-defined cell identities. Once the cells have been labelled using the clustering algorithm, cell generation distributions are estimated on the basis of the labelled lineage trees.
-
A second one that favours the temporal structuring: In this approach, the data of interest are lineage forest and the only spatial structuring taken into account corresponds to siblings with respect to a given parent cell. In a first step, cell identities are inferred on the basis of the cell descriptors taking into account lineage relationships using hidden Markov tree models and the spatial regions that emerge from the cell identity labelling are then characterized. This second approach is supported by the fact that cell topology is only affected by division which makes highly relevant the local spatial information taken into account in this approach.
Mechanical models of plant tissues
Participants : Jean-Philippe Bernard, Olivier Ali, Christophe Godin, Benjamin Gilles, Frédéric Boudon, Ibrahim Cheddadi, Jan Traas [ENS-Lyon] , Olivier Hamant [ENS-Lyon] , Arezki Boudaoud [ENS-Lyon] .
This research theme is supported by the Inria Project Lab Morphogenetics and the Jan Traas's ERC.
The rigid cell walls that surround plant cells are the main load-bearing structures in plant tissues. These walls are submitted to stresses due to cell turgor pressure. Above some threshold, these stresses cause deformation in the cell walls and triggers wall irreversible expansion (synthesis). Shape changes of plant tissues are therefore tightly related to the turgidity of cells and to the mechanical state and the molecular composition of the underlying cell walls. We developped a conceptual and numerical framework to model the mechanical structure of cell walls and their deformation by turgor pressure in 3-dimensions. This framework was used to study the interplay between post-transcriptional regulation, biochemistry, and mechanics within growing plant tissues. This work has been published this year in Plos Computational Biology [13] .
In this first step, all mechanical and structural quantities are defined at the tissular scale. This is made possible by abstracting the connection between the actual molecular composition of the walls and the various signalling cascade at play during growth. To extend this approach, we also started to develop a mechanobiological approach relating the irreversible expansion of the walls to molecular mechanisms happening within them, based on the thermodynamical equilibrium of the pectin-based matrix within the wall. We propose that at the molecular scale expansion of this matrix is based on the adsorption of newly synthetized pectin molecules. This adsorption mechanism is regulated by the mechanical stresses applied on the wall. We show that this mechanism belongs to a class of biochemical / biomechanical processes commonly appearing in the dynamics of supra-molecular load-bearing structures: the force-driven polymerization processes. A preliminary version of these ideas (the 1D case) is currently under review in Trends In Plants Sciences.
We also considered to extend the original modeling approach to situations where entire organ dynamics should be modeled over large time lapse (several days) (PhD work of Jean-Philippe Bernard). In our first approach, the mechanical model relies on a finite element method (FEM) to describe the deformation of the tissue. In FEM, the tissue is represented by a mesh. The positions of the vertices at each time step are estimated from a linear system. If the tissue is big or if the mesh is fine, the linear system can be large and thus leads to computational overheads. An alternative way to classical FEM is to use a meshless method where the deformation of the tissue can be characterized by a linear combination of deformations of a finite and small set of frames. Because shape functions are no longer defined on each element but on the whole tissue, they have to be updated at each growth step by estimating a new rest configuration. With meshless method, the discretization of the system can be dynamically updated parsimoniously according to the precision required to model the emergence of shapes. With an uniform distribution of the frames within the volume, our method still leads to computational overheads. However, since the meristem initiates a branching structure at a macroscopic scale, we combined our mechanical model at tissular resolution with classical method used to generate branching structures at macroscopic scales. For this, we use the information of the plant branching structure to distribute the frames along the plant's axes. This allows us to use curvilinear shape functions while describing the branching structure growth using L-systems. This multi-scale framework allows us to define developmental rules which can initiate new organs at the surface of the meristematic dome by softening locally the meristem dome and thus creatin new growing initia. First very encouraging results were obtained this year that demonstrate the feasibility of the approach.
Gene regulatory networks: Design of a genetic model of inflorescence development.
Participants : Eugenio Azpeitia, Christophe Godin, François Parcy, Etienne Farcot.
This research theme is supported by the Inria Project Lab Morphogenetics.
Modeling gene activities within cells is of primary importance since cell identities correspond to stable combination of gene expression.
We studied the regulatory network that controls the flowering transition during morphogenesis. To overcome the network complexity and integrate this regulation during ontogenesis, we have developed a first model of the control of floral initiation by genes, and in particular the situation of cauliflower mutants, in which the meristem repeatedly fails in making a complete transition to the flower. Three different network models were done and validate. A first Boolean version, a second fuzzy logic and an ODEs models were studied. The models are able to correctly recover the gene steady states observed in the meristems during the flower transitions, the gene transitions and the mutant effects. Importantly, the model is able to explain the cauliflower mutants. This work couples models at different scales, since the gene regulatory network is used as a decision module in an L-system model of the inflorescence architecture. This mixed model has led us to make different hypotheses about gene interactions and hormonal regulation. First predictions about gene actors controlling the passage to flower could be verified. Some links between gene regulation and plant growth have been identified. These links can be experimentally tested which could lead to a first integrated picture of flower development.
Finally, given that the cauliflower have different morphologies (i.e. regular and romanesco cauliflower morphologies) we explored the effect of changes in the L-system parameter values over the cauliflower morphology. Interestingly, we discovered by exploring the model that variations in the regulation of some phyllotactic parameters can produce the different cauliflower morphologies and explain other reported differences among them. Predictions were made using the model and experimental validations of this hypothesis are curren being tested. All our results could provide a comprehensive understanding of how does genes and plant architecture are linked in a dynamical way.
Modelling the influence of dimerisation sequence dissimilarities on the auxin signalling network
Participants : Jonathan Legrand, Yann Guédon, Teva Vernoux [ENS-Lyon] .
Auxin is a major phytohormone involved in many developmental processes by controlling gene expression through a network of transcriptional regulators. In Arabidopsis thaliana, the auxin signalling network is made of 52 potentially interacting transcriptional regulators, activating or repressing gene expression. All the possible interactions were tested in two-way yeast-2-hybrid experiments. Our objective was to characterise this auxin signalling network and to quantify the influence of the dimerisation sequence dissimilarities on the interaction between transcriptional regulators. We applied model-based graph clustering methods relying on connectivity profiles between transcriptional regulators. Incorporating dimerisation sequence dissimilarities as explanatory variables, we modelled their influence on the auxin network topology using mixture of linear models for random graphs. Our results provide evidence that the network can be simplified into four groups, three of them being closely related to biological groups. We found that these groups behave differently, depending on their dimerisation sequence dissimilarities, and that the two dimerisation sub-domains might play different roles. We proposed the first pipeline of statistical methods combining yeast-2-hybrid data and protein sequence dissimilarities for analyzing protein- protein interactions. We unveil using this pipeline of analysis the transcriptional regulator interaction modes.
Model integration
Participants : Frédéric Boudon, Christophe Godin, Guillaume Baty, Guillaume Cerutti, Jean-Louis Dinh, Jan Traas.
This research theme is supported by the Morphogenetics Inria Project Lab.
Our approach consists of building a programmable tissue which is able to accept different modeling components. This includes a central data structure representing the tissue in either 2-D or 3-D, which is able to grow in time, models of gene activity and regulation, models of signal exchange (physical and chemical) between cells and models of cell cycle (which includes cell division). An introduction to the modeling of some main components of such integrated system was published as a book chapter in the series of Ecole de Physique des Houches [43] . For each modeling component, one or several approaches are investigated in depth, possibly at different temporal and spatial scales, using the data available from the partners (imaging, gene networks, and expression patterns). Approaches are compared and assessed on the same data. The objective of each submodel component will be to provide plugin components, corresponding to simplified versions of their models if necessary, that can be injected in the programmable tissue platform. This work is developed in collaboration with the RDP group at ENS-Lyon [70] and the CPIB group in Nottingham, UK [53] .
One key aspect of our approach is the development of a computer platform dedicated to programming virtual tissue development, TissueLab. This platform, based on OpenAlea, will be used to carry out integration of the different models developed in this research axis. In the past year, progress has been made in defining a generic tissue data structure that could be used in this platform. Currently, robust geometric operations such as division are implemented and tested. Moreover, a redesign of the structure based on more elaborated formalisms such as combinatorial maps is being investigated. A 2D version is being developed in the context of Jean-Louis's Dinh PhD thesis, and will be described in a forthcoming book chapter.